|
The "Holy Grail" of computational chemistry is the calculation of the
molecular orbitals (MOs) for a given molecule. IF we can calculate
the MOs for a molecule, THEN we can know lots of things about the
molecule, including its:
- energy
- electron density
- electrostatic potential
- transition state (if any)
- frequency
However, calculating MOs is not that easy. The computer, however, does this
for us. But, you, the chemist, must tell the computer some information
first. Input files for all of the major computational chemistry
packages contain these three parts:
- Geometry which includes
- Bond lengths
- Bond Angles
- Dihedrals
- Kind of Calculations:
- Single point energy
- Frequency
- Transition state
- Electron density
- Electrostatic potential
- Starting set of mathematics and approximations
- Calculation method (molecular mechanics, semiempirical, or ab initio
- Type(s) of approximation (Hartree-Fock, Moller-Plesset, etc.)
- Basis Set Approximation
The graphic below "captures" the essence of what is the responsibility of the
chemist and what is the responsibility of the computer:
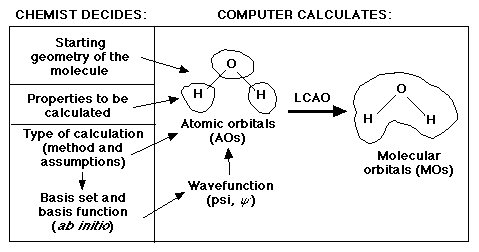
In this lab we will be looking at the third ingredient, basis sets.
A molecular-orbital theory calculation is a mathematical expression of an
electron in a molecule. Although there are many types of molecular-orbital
functions, in this lab we will only look at the Slater Type Orbitals (STOs)
and the Gaussian Type Orbitals (GTOs).
Although there is not a major difference in these two methods when
calculating small molecules, major discrepencies arise for larger molecules of
30 or more atoms. STOs require more calculating, which takes tremendous
amounts of time, however their calculations have been found to be more
accurate than GTOs. On the other hand, GTOs, although less accurate, are much
faster to calculate than STOs. This forced scientists to compromise time or
accuracy.
Eventually, scientists realized that by adding several GTOs, they were able
to mimic the STOs accuracy. In fact, as the number of GTOs used increased, the
better they were able to model the STO equation.
When using GTOs to model STOs, the new equations are given a new name. They
are identified as STO-kG equations where k is a constant that represents the
number of GTOs used. For instance, two common equations are the STO-3G and the
STO-6G in which 3 and 6 GTOs are used respectively.
|