ChemVizLab Activities |
Geometry Optimization Lab: Additional Background | |||||||||
| Geometry Optimization Lab Activity
Students
TeachersCartesian Converter Materials Readings Lab Activities Support Materials
Related Links Developers' Tools
|
The geometry of a molecule determines many of its physical and chemical
properties. This is why it is very important that we understand the geometry
of a molecule when running computations. In computational chemistry we are
specifically concerned with optimizing:
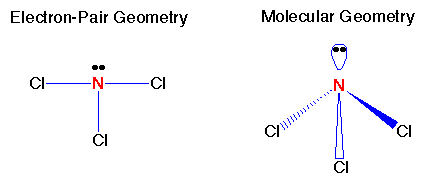
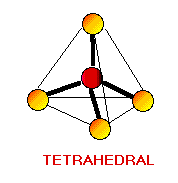
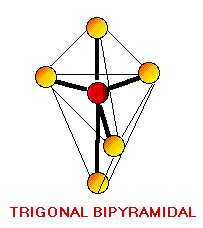
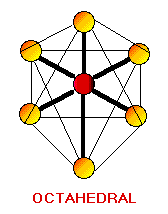
Remember the VSEPR (valence shell electron-pair repulsion) model. This model was designed by Ronald J. Gillespie. It states that the geometry of a molecule is determined by the repulsion forces of its valence electron pairs. At this time, it is important to remember that not all valence electrons are bonded. Electrons which are not bonded are called lone pairs. Lone pairs have the same effect as bonded electron pairs. The repulsion forces of the valence electrons directly effect the size of the bond angle. The bond angle is the angle formed by two pairs of valence electrons and the central atom that connects the two. The stronger the repulsion strength, the larger the bond angle. The strength of repulsion goes in order: lone pair - lone pair > lone pair - bond pair > bond pair - bond pair Below are diagrams of various geometries as predicted by the VSEPR model.
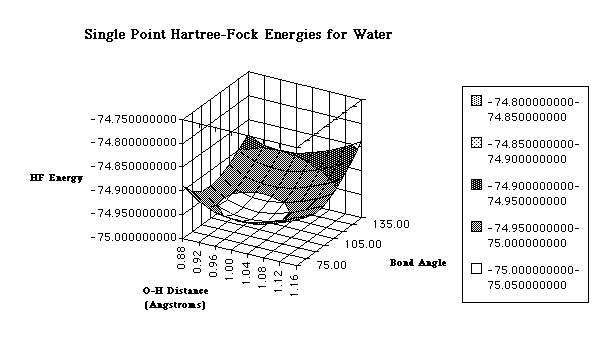
 Although some textbooks choose to ignore the length of bonds in their geometry calculations, the bonds also have an effect on the geometry of the molecule. In fact, as you will soon see, their effect will be crucial in our computational geometry optimizations. Now, up to this point, the determination of a molecule's geometry has been fairly simple: identify the valence electron pairs and determine the geometry. However, the VSEPR model is only a visual model and does not give us the detail needed in computational chemistry. For computational chemistry we need to be more precise by using cartesian coordinates, bond lengths and bond angles to find the optimal molecular geometry. The arrangement of atoms in the molecules and more specifically the electrons around the atom determine the energy level of that molecule. In fact, the energy of a molecular system varies even with small changes in its structure. This is why geometry is so important when performing calculations. The objective of a geometry optimization is to find the point at which the energy is at a minimum because this is where the molecule is most stable and most likely to be found in nature. One way to observe the effect of different geometries on energy level is to calculate a potential energy surface (PES). The potential energy surface is just a mathematical relationship correlating the particular molecular structure and its single point energy. They are usually represented by three-dimensional plots, although they can also be simple x-y plots for diatomic molecules. Potential energy surfaces are characterized by distinct points:
 In most programs that calculate geometry optimizations, the user specifies a beginning geometry (an educated guess), either as internal coordinates (Z-matrix), Cartesian coordinates, or in mixed format. Then a basis set is specified, the program will then compute the energy and the gradient at that point, decides if it has reached a stopping point (convergence), and then varies the geometry based on the size of the gradient. New integrals are calculated, new self-consistent field calculations are done, and a new energy and gradient are calculated. These steps are repeated until the program reaches convergence i.e. finds a stationary point. There are a number of different algorithms for performing optimizations, such as Berny, Fletcher-Powell, quasi-Newton, and others. Most of these optimization algorithms also calculate the second derivative of the energy with respect to the coordinates, known as a Hessian. The Hessian serves to specify the curvature of the surface for that particular geometry, and thus "optimizes" the determination of how to vary the geometry for the next step. There are also things that the computational chemist can do to make optimizations behave better such as:
|
in cooperation with the
National Center for Supercomputing Applications
© Copyright 1999-2000 The Shodor Education Foundation, Inc.