|
Home
Geometry Optimization Lab Activity
Geometry Optimization Main Page
Students
Introduction
Objectives
Background Reading
Procedure
Questions
Further Work
References/Support Materials
Teachers
Additional Background
Materials
Standards
First year chemistry curriculum concepts
Second year chemistry curriculum concepts
Cartesian Converter Materials
Z-matrix to Cartesian Converter
Cartesian Converter Example
Help Instructions for the Z-matrix to Cartesian Converter
Readings
Overview
Atomic Orbitals
Lab Activities
Z-matrices
Basis Sets
Geometry Optimizations
Ionization Energies
Support Materials
Interactive Tools
Glossary of Terms
Quick Guide to DISCO Output File
Related Links
ChemViz
Computational Chemistry
SUCCEED's Computational Chemistry
Developers' Tools
What's New?
Discussion Board
Team Members
Email the Group
Contact Webmaster
|
Key Points
You have learned in earlier sessions that the geometry of a molecule
determines many of its physical and chemical properties. This is why having
accurate molecular geometries is so important to scientists. When calculating
a molecular geometry, there are three measurements that we can make:
The objective of geometry
optimization is to find an atomic arrangement which makes the molecule
most stable. Molecules are most stable when their energy is low. So, in order
to optimize a molecular geometry, you want to test various possibilities to
see which one has the lowest energy value. One way scientists do this is by
creating a Potential Energy Surface
(PES). A PES is a mathematical relationship between different molecular
geometries and their corresponding single
point energies. These values are usually displayed in a three-dimensional
graph (although simple x-y plots are sometimes used for diatomic
molecules). The three dimensions represent the bond angle, bond distance and
the Hartree-Fock energy value. Below is
an example of the PES of water.
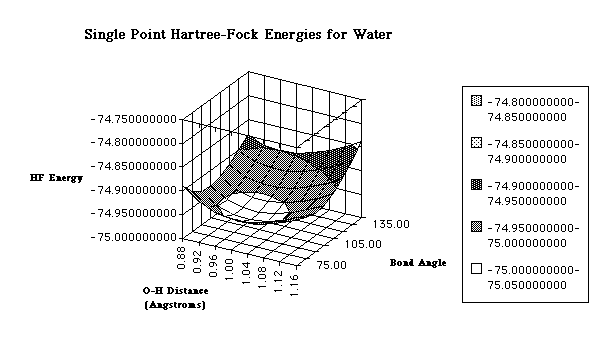 Potential energy surfaces are characterized by distinct points:
Potential energy surfaces are characterized by distinct points:
- Local Maximum - that point on the
potential energy surface that is the highest value in a particular section or
region of the PES
- Global Maximum - that point on the
potential energy surface that is the highest value in the entire PES
- Local Minima - that point on the
potential energy surface that is the lowest value in a particular section or
region of the PES
- Global Minima - that point on the
PES that is the highest value in the entire PES
- Saddle point - a point on the PES
that is a maximum in one direction and a minimum in the other. Saddle points
represent a transition structure connecting two equilibrium structures.
Below is a two dimensional graph so that you can clearly observe the various
points. (A saddle point is not represented in this picture because it can only
be shown in a three dimensional graph.)
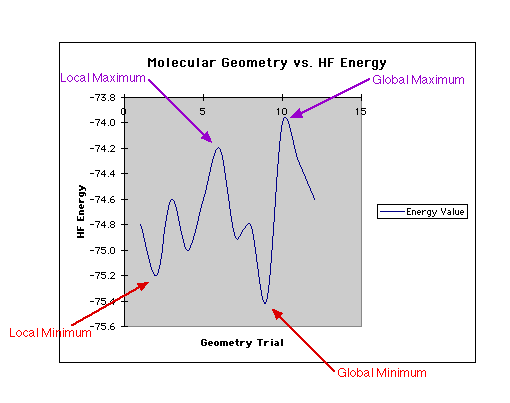 Notice that a global minima represents the most optimal molecular geometry.
The PES gives us a quick way to look and approximate a reasonable geometry.
Notice that a global minima represents the most optimal molecular geometry.
The PES gives us a quick way to look and approximate a reasonable geometry.
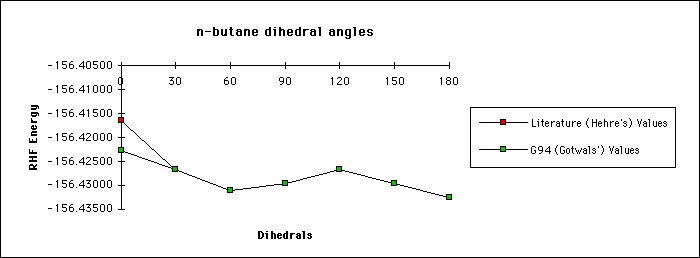
To really understand the effect that geometry has on single point energy
calculations, consider the example of n-butane below. In this example we
compare literature values for single point energies with experimental
values. More importantly, we compare single point energy calculations for
various sized dihedrals. Notice how the energy values change as the dihedral
grows.
Data Results:
CCCC Dihedral Literature (Hehre's) Values G94 (Gotwals') Values
0 -156.41638 -156.42272
30 -156.42670 -156.42666
60 -156.43106 -156.43106
90 -156.42958 -156.42956
120 -156.42673 -156.42667
150 -156.42963 -156.42962
180 -156.43247 -156.43247
Graphical Results:
Most computational chemistry programs optimize molecular geometries for
you. They do this by the series of steps that you read about in the Introduction. Remember that you typically must give
the program an initial geometry and a basis set. From there the program
calculates optimal values.
|